- I. Connection au serveur ESILBAC et pratique de LINUX
- II. Rechercher les séquences des génomes de Listeria dans GENBANK
- III. Manipuler les fichiers de séquences
- IV. Rechercher les gènes dans les séquences avec EMBOSS
- V. Rechercher les "vrais" gènes avec BLAST
- VI. Trouver de l'information sur les génomes avec GENBANK
- VII. Rechercher les gènes de virulence avec Artemis ACT
- VIII. Visualisation de la structure 3D des protéines sélectionnées avec PDB et PyMol
- Guide de survie dans le terminal
I. Connection au serveur ESILBAC et pratique de LINUX
Branchement à SIM
- Se brancher à SIM (PC Windows) au M-6
Branchement aux serveurs ESILBAC via Hummingbird
- Double-cliquer sur le répertoire Logiciels spécialisés situé sur le bureau
- Double-cliquer sur le répertoire Hummingbird
- Choisir xdm_esilbac1 ou xdm_esilbac2
- Inscrire son code d'accès et son mot de passe
Linux
Pratiquons quelques commandes de LINUX
La casse est importante : 'monfichier.txt' est différent de 'Monfichier.txt'
Ne pas utiliser de caractères spéciaux : accent, espace, ponctuation, ...
Les commandes se valident avec la touche [ Enter ]
- Voir la liste des fichiers dans un répertoire
ls: list (engl) = dresser une liste (fr)ls
- Créer un répertoire
mkdir : make directory (engl) = créer un répertoire (fr)mkdir sejour_decouverte
- Visiter un répertoire
cd: change directory (engl) = se déplacer dans l'arborescence (fr)cd sejour_decouverte
- Monter dans un répertoire
cd ..
- Déterminer l'endroit où l'on se trouve dans l'arborescence
pwd: print working directory name (engl) = inscrire le nom du répertoire à l'écran (fr)pwd
- Compléter une commande, un nom de fichier lorsqu'on en tape les premières lettres
Touche tab
II. Rechercher les séquences des génomes de Listeria dans GENBANK
GENBANK est une banque de données de séquences génétiques reconnue internationalement. Elle est maintenue aux Etats-Unis par le NCBI. On y trouve plus de 108 millions de séquences d'ADN publiquement disponibles. Le NCBI met également à la disposition de la communauté internationale de nombreux outils d'analyse de génome et un serveur de fichiers (FTP).
- Ouvrir un navigateur (firefox, ...)
- Dans une fenêtre de terminal, en utilisant la commande suivante:
firefox &
- OU via l'interface graphique en cliquant sur l'icone du programme
- Dans une fenêtre de terminal, en utilisant la commande suivante:
- Se connecter sur le site ftp de GENBANK dédié aux bactéries en tapant l'adresse suivante dans le navigateur:
ftp://ftp.ncbi.nih.gov/genomes/Bacteria - Cliquer sur le dossier Listeria_innocua
Cliquer avec le bouton droit de la souris sur les fichiers suivants et enregistrer sous format texte (Enregistrer la cible du lien sous ou save link target as ou save link as) dans votre répertoire en les renommant comme ci-dessous :- NC_003212.faa renommé en lin_genes_prot.txt
- NC_003212.ffn renommé en lin_genes_dna.txt
- NC_003212.fna renommé en lin_genome_dna.txt
- Revenir en arrière et cliquer sur le dossier Listeria_monocytogenes
Cliquer avec le bouton droit de la souris sur les fichiers suivants et enregistrer sous format texte (Enregistrer la cible du lien sous ou save link target as ou save link as) dans votre répertoire en les renommant comme ci-dessous :- NC_003210.faa renommé en lmo_genes_prot.txt
- NC_003210.ffn renommé en lmo_genes_dna.txt
- NC_003210.fna renommé en lmo_genome_dna.txt
III. Manipuler les fichiers de séquences
Nous allons afficher successivement dans le terminal le contenu des trois fichiers pour Listeria monocytogenes (lmo) en utilisant le programme more qui sert à afficher les premières lignes d'un fichier.
Dans une fenêtre de terminal :- Afficher les premières lignes du génome de Listeria monocytogenes en tapant la commande suivante :
more lmo_genome_dna.txt
- La barre [ Espace ] affiche la suite du fichier
- La touche [ q ] permet de quitter le programme
Remarquer le format particulier de ce fichier avec la première ligne commençant par un > ; il s'agit d'un fichier en format fasta - Afficher le fichier contenant les gènes de Listeria monocytogenes
more lmo_genes_d2na.txt
Remarquer que les séquences nucléiques des gènes sont en format fasta - Afficher le fichier contenant les protéines de Listeria monocytogenes
more lmo_genes_prot.txt
Remarquer que les séquences protéiques des gènes sont en format fasta - Maintenant, nous allons compter le nombre de gènes dans le génome de Listeria
monocytogenes. Pour ce faire nous allons tirer parti du format fasta et de la puissance de
l'outil grep. Ce dernier cherche dans un fichier spécifié un patron demandé (un mot,
une phrase, un caractére, etc.).
grep -c '>' lmo_genes_dna.txt
-c : cette option donne comme résultat le nombre de lignes qui vérifient la recherche
'>' : patron, motif
lmo_genes_dna.txt : fichier d'entrée dans lequel s'effectue la recherche - Compter le nombre de gènes dans le génome de Listeria innocua
grep -c '>' lin_genes_dna.txt
- Qui a le plus de gènes ?
- Peut-on en déduire quelle Listeria est dangereuse ?
IV. Rechercher les gènes dans les séquences avec EMBOSS
EMBOSS est une suite logicielle d'analyse de séquences. Cette suite logicielle est disponible gratuitement et la plupart des institutions de recherche l'installent sur leur serveur. Les logiciels sont lancés en ligne de commande (dans une fenêtre de Terminal accédant au serveur). Il existe également des interfaces graphiques comme celle mise en place sur le serveur du centre Robert Cedergren : http://anabench.bcm.umontreal.ca/html/EMBOSS/
Nous allons rechercher dans une portion de la séquence du génome de Listeria monocytogenes tous les gènes potentiels. On utilisera une règle simple (cf ci-dessous). Cette règle ne permet pas de trouver la totalité du gène (qui commence un peu avant et se termine après) mais la partie qui donnera la protéine. Cette portion est appelée ORF pour "Open Reading Frame" (phase ouverte de lecture) ou CDS pour "Coding sequence" ( séquence codante) lorsque la structure du gène est confirmée.
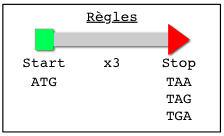 |
 |
- Charger EMBOSS
module load emboss
- Visualiser la liste des programmes disponibles dans EMBOSS
wossname
Cliquer deux fois sur la touche [ Enter ]
La liste des programmes disponibles dans EMBOSS apparait, triée par catégories.
Dans la catégorie EDIT, se trouve le programme extractseq pour extraire une portion de la séquence d'ADN.
Dans la catégorie NUCLEIC GENE FINDING se trouve le programme plotorf qui recherche les ORFs. - Extraire, avec le programme extractseq (EDIT) sla sous-séquence de 450 000 à 460 000 chez Listeria monocytogenes et la copier dans le fichier lmo_genome_450-460_dna.txt
extractseq lmo_genome_dna.txt -regions 450000-460000 -outseq lmo_genome_450-460_dna.txt
lmo_genome_dna.txt : fichier d'entrée
-regions 450000-460000 : on extrait la portion 450000 à 460000
-outseq lmo_genome_450-460_dna.txt fichier de sortie qui contient les 10 000 bases extraites - Utilisons le programme plotorf (NUCLEIC GENE FINDING) en mode interactif
plotorf
- Indiquer le nom du fichier contenant la sous-séquence de Listeria monocytogenes (lmo) lorsque le programme vous le demande
Input sequence : lmo_genome_450-460_dna.txt
- Indiquer le format de sortie
Graph type [x11]: png
- Le programme indique qu'il a créé le fichier de sortie plotorf.1.png. Pour visualiser ce fichier, utiliser la commande display
display plotorf.1.png
- Que représentent les barres bleues ?
- Que représentent les six pistes nommées F1, F2, F3, R1, R2, R3 ?
- Sachant qu'un gène bactérien fait en moyenne 1000 bases, combien de gènes devrait-on trouver ?
- Que pensez-vous du résultat obtenu ?
V. Rechercher les "vrais" gènes avec BLAST
BLAST signifie Basic Local Alignment Search Tool. Cet outil cherche des régions de similarité locale entre les séquences. Le programme compare les séquences de nucléotide ou de protéine à d'autres séquences contenues dans une énorme base de données et calcule des statistiques de significativité
Notre programme a prédit beaucoup de gènes dont beaucoup sont chevauchants. Nous allons extraire toutes les protéines traduites à partir des ORFs prédites et rechercher s'il existe déjà des gènes similaires connus.
- Dans une fenêtre de terminal : créer un fichier avec toutes les protéines traduites à partir des ORFs trouvées avec la commande getorf
getorf lmo_genome_450-460_dna.txt -outseq lmo_genome_450-460_ORF_prot.txt -find 1
lmo_genome_450-460_dna.txt : fichier d'entrée qui contient la séquence dans laquelle on va chercher les ORFs
-outseq lmo_genome_450-460_ORF_prot.txt : fichier de sortie qui contient les ORFs protéiques
-find 1 option pour traduire les ORFs en protéines - Dans un navigateur (firefox, ...) : aller sur la page BLAST du NCBI
http://blast.ncbi.nlm.nih.gov/
Sur la page qui s'affiche:- Dans la section Basic BLAST, cliquer sur le lien protein BLAST
- Dans la section Enter Query Sequence, cliquer sur [ Browse ] pour charger le fichier lmo_genome_450-460_ORF_prot.txt
- Dans la section Choose Search Set
- Pour le paramètre Organism, indiquer la valeur 2, cela limitera la recherche de protéines similaires aux bactéries
- Pour l'option Exclude, cocher Uncultured/environmental sample sequences, pour accélérer la recherche
- Dans la section Algorithm parameter (sous le gros bouton bleu [ BLAST ])
Cliquer sur la flèche pour afficher le contenu- Dans la section General Parameters, pour le paramètre Max target sequences, changer la valeur à 10 pour ne pas être submergé par les résultats
- Cliquer sur le bouton bleu [ BLAST ] pour lancer l'outil
La recherche peut prendre plusieurs minutes
La liste des ORFs que nous avons soumise en entrée est dans le menu déroulant. Les ORFs pour lesquelles aucune similarité n'est trouvée sont grises, les autres sont en noir.
- Pourquoi ne trouve-t-on pas de similarité pour certaines ORFs?
- Que remarque-t-on concernant le nombre d'ORFs sans similarité par rapport au nombre d'ORFs avec ?
- Sélectionner le premier résultat positif, la ligne 8 correspondant à l'ORF de 386 acides aminés (386aa). Pourquoi trouve-t-on des similarités chez d'autres espèces ?
- Quelle règle simple pourrait-on trouver pour éviter toutes ces "fausses ORFs" dans nos recherches de gènes?
VI. Trouver de l'information sur les génomes avec GENBANK
Le processus qui consiste à rechercher les (vrais) gènes sur un génome et leur associer une fonction biologique est l'annotation.
L'annotation des deux génomes de Listeria qui nous intéressent a déjà été réalisée par des experts de cette espèce. Lorsque l'annotation a été terminée, les équipes de recherche ont publié un article scientifique pour présenter leur travail et ont déposé l'annotation dans les banques de données publiques dont GENBANK fait partie. Nous allons rechercher les informations sur l'annotation de ces bactéries.
Dans un navigateur (firefox, ...)- Allez sur le site Entrez via l'adresse http://www.ncbi.nlm.nih.gov/Entrez/
- Cliquer sur Genome : whole genome sequences dans la colonne de gauche
- Dans la zone de recherche Search for indiquer Listeria[orgn], puis cliquer sur [ Go ]
- Sélectionner les items 5 et 6 : NC_003212 et NC_003210
- En haut de la page, dans le menu Display, sélectionner Overview
La page se recharge automatiquement en affichant les informations pour les deux organismes sélectionnés
- Quelles sont les informations que l'on apprend sur les deux organismes ?
- Cela nous aide-t-il pour savoir si l'un des deux est pathogène ?
- Et en cliquant sur le lien Genome project de la colonne Link du tableau
VII. Rechercher les gènes de virulence (dangereux) avec Artemis ACT
Artemis ACT est un programme de visualisation de comparaison de génomes développé au Sanger Institut. Une version "Java Web Start" de cet outil est disponible sur le site web au JGI (Joint Genome Institut) de Californie avec les comparaisons entre génomes pré-calculées.
Dans un navigateur (firefox, ... )
- Aller sur le site des ressources en microbiologie (IMG : Integrated Microbial Genomes) du
JGI
http://img.jgi.doe.gov/ - Dans le menu en haut de la page, cliquer sur Compare Genomes
- Dans le sous-menu qui s'est affiché, cliquer sur Synteny viewers.
- Une liste d'outils s'affiche, cliquer sur Artemis-ACT.
- Sur la page ACT Genome Selection, sélectionner (en maintenant la touche Ctrl) les
espèces : Listeria innocua Clip 11262 et Listeria monocytogenes EGD-e.
Cliquer sur [ Next ] - Sur la page Pairwise selection, cliquer sur [ Next ].
- Sur la page Contig Reorder - Artemis - ACT,
Dans la colonne Ignore du 2e tableau, cocher la seconde ligne :
2 NC_003383 Listeria innocua Clip11262 plasmid pLI100: NC_003383
Ceci écartera le plasmide de l'analyse
Cliquer sur [ Next ].
Le programme lance des processus ... - Lorsqu'une nouvelle page intitulée Artemis - ACT s'affiche, cliquer sur le bouton [ Run ACT ] qui lancera une application en Java Web Start.
- L'application ACT affiche en haut de la page, le génome de Listeria innocua et en bas
celui de Listeria monocytogenes avec leurs annotations dans les 6 phases de lecture.
S'il existe de fortes similarités entre deux portions de génome, une barre rouge les relie.
Les barres de défilement horizontales tout en haut et tout en bas de la fenêtre permettent de se déplacer sur le génome. Le déplacement est coordonné entre les deux génomes.
Utiliser les barres de défilement horizontal afin d'afficher à l'écran la portion de génome sur laquelle nous avions recherché les ORFs, c'est à dire la portion 450000 à 460000 sur le génome de Listeria monocytogenes.
- Est-ce que nous retrouvons tous les gènes que nous avions prédits (après BLAST) ?
- Sur quel critère peut-on se baser pour trouver les gènes de virulence (qui rendent la bactérie dangereuse, pathogène) ?
- Quels sont les gènes qui pourraient être des gènes de virulence ?
VIII. Visualisation de la structure 3D des protéines sélectionnées avec PDB et PyMol
La PDB (Protein Data Bank) est une banque de données de structure tridimensionnelle de molécules.
Dans un navigateur (firefox, ... )
- Se connecter sur le site de la PDB
http://www.pdb.org/ - Dans le champs texte de recherche en haut de page, indiquer 2OMT (la lettre O)
Puis, cliquer sur le bouton Search
La fiche de la structure cristallographique de la protéine sélectionnée et de la Cadherine, son récepteur sur les cellules humaines s'affiche. - Dans le menu à droite, choisissez Download Files puis PDB File (Text).
Le fichier est sauvé sous le nom 2OMT.pdb. - Nous allons utiliser le programme de visualisation moléculaire Pymol afin de l'observer en 3D.
Dans une fenêtre de terminal , taperpymol
- Dans la petite fenêtre grise, faites File > Open pour ouvrir le fichier 2OMT.pdb
Puis, dans la fenêtre Pymol viewer, dans le menu à droite, sélectionner :- S(show) et cartoon
- H(hide) et lines
- H(hide) et waters
- C(color) spectrum et rainbow
- Saurez-vous reconnaitre laquelle de ces deux molécules est la Cadherine ?
L'annotation et le métier de bio-informaticien
La conception d'outils pour la recherche de gènes et la manipulation de séquences requiert de bonnes connaissances en algorithmique et en statistique. Ces outils doivent être rapides et donner des résultats pertinents.
L'implémentation des outils afin de les rendre "facilement" utilisables nécessite des aptitudes en programmation et en interface homme/machine.
Bien qu'une partie du travail de l'annotateur soit facilitée par l'amélioration des programmes de recherche de gènes et l'automatisation de suite de processus, l'annotation est une tâche longue qui requiert beaucoup de rigueur et une bonne connaissance de la biologie des organismes.
Guide de survie dans le Terminal
NOTES
------
1 / La touche [ TAB ] permet de completer une commande, un nom de fichier lorsqu'on en tape les premieres lettres
2 / Il faut taper sur la touche [ Entrer ] a la fin de chaque commande pour la valider
3 / La casse compte : 'monfichier.txt' est different de 'Monfichier.txt'
4 / Utiliser les fleches haut et bas pour se deplacer dans l'historique de commande
!!!!! Le signe '>' indiquera qu'il faut taper la commande qui le suit ; il ne faut pas l'ecrire !!!!!!!
ARBORESCENCE
------------
/ C'est le repertoire le plus bas, la "racine"
|------ A
|------ a
|------ b
|------ fichier1.txt
|------ B
Pour indiquer un chemin, on separe les repertoires par des "/".
Par exemple, le chemin depuis la racine jusqu'au "fichier1.txt" est : /A/b/fichier1.txt
OU SUIS-JE ?
-------------
> pwd Position dans l'arborescence des fichiers
> cd Revenir dans son repertoire initial ("home")
> cd monrepertoire/ se deplace dans le sous-repertoire "monrepertoire"
> cd .. Remonte d'un repertoire
> cd ../.. Remonte de 2 repertoires
> ls Fait la liste du contenu du repertoire courant (en lignes)
> ls -l Fait la liste du contenu du repertoire courant (sous forme de liste)
FICHIER
--------
> touch toto.txt Cree le fichier toto.txt (dans le repertoire dans lequel on se trouve)
> more toto.txt Affiche le contenu du fichier toto.txt par page
[ Espace ] pour faire defiler le fichier
[ q ] pour quitter
> cat toto.txt Affiche TOUT le contenu du fichier toto.txt. Attention aux fichiers tres longs !
> rm toto.txt Supprime le fichier toto.txt
A la demande de confirmation, taper [ y ] pour valider
REPERTOIRE
----------
> mkdir tata Cree le repertoire "tata" dans le repertoire dans lequel on se trouve
KES C CA ?
----------
> man macommande Affiche le manuel pour la commande "macommande".
Ex: > man more
AIE AIE AIE
-----------
ctrl + c *Arreter* la commande en cours
ctrl + z *Interrompre* la commande en cours
> fg pour la relancer
> bg pour la relancer en arriere plan
COMMANDES AVANCEES
---------------------
> grep "motif" fichier *Affiche* les lignes contenant "motif" dans "fichier"
Ex : grep "ribosomal" lmo_genes.dna Recherche les lignes du fichier "lmo_genes.dna" qui contiennent le mot "ribosomal"
> grep -c "motif" fichier *Compte* les lignes contenant "motif" dans "fichier"